
Апоптоз сердечная недостаточность
Апоптоз — запрограммированная гибель клетки
Что такое апоптоз?
Апоптоз – физиологическая смерть клетки, представляющая собой своеобразную генетически запрограммированную самоликвидацию.
Термин «апоптоз» в переводе с греческого означает «опадающий». Авторы термина дали такое название процессу запрограммированной смерти клеток потому, что именно с ним связано осеннее опадание увядших листьев. Кроме того, само название характеризует процесс как физиологический, постепенный и абсолютно безболезненный.
У животных в качестве наиболее яркого примера апоптоза, как правило, приводят исчезновение хвоста у лягушки во время метаморфозы из головастика во взрослую особь.
По мере взросления лягушонка хвост полностью исчезает, поскольку его клетки подвергаются постепенному апоптозу – запрограммированной смерти, и поглощению деструктированных элементов другими клетками.
Явление генетически запрограммированной гибели клеток встречается у всех эукариотов (организмов, клетки которых имеют ядро). Прокариоты же (бактерии) имеют своеобразный аналог апоптоза. Можно сказать, что данный феномен характерен для всего живого, за исключением таких особых доклеточных форм жизни, как вирусы.
Апоптозу могут подвергаться как отдельные клетки (как правило, дефектные), так и целые конгломераты. Последнее особенно характерно для эмбриогенеза. К примеру, опыты исследователей доказали, что благодаря апоптозу во время эмбриогенеза исчезают перепонки между пальцами на лапках у цыплят.
Ученые утверждают, что у человека такие врожденные аномалии, как сросшиеся пальцы на руках и ногах, также возникают вследствие нарушения нормального апоптоза на ранних стадиях эмбриогенеза.
История открытия теории апоптоза
Изучение механизмов и значения генетически программируемой клеточной смерти началось еще в шестидесятых годах прошлого века. Ученых заинтересовал тот факт, что клеточный состав большинства органов на протяжении жизни организма практически одинаков, а вот жизненный цикл различных типов клеток значительно отличается. При этом происходит постоянная замена многих клеток.
Таким образом, относительное постоянство клеточного состава всех организмов поддерживается динамическим равновесием двух противоположных процессов – клеточной пролиферации (деление и рост) и физиологического отмирания отживших клеток.
Авторство термина принадлежит британским ученым – Дж. Керру, Э. Уайли и А. Керри, которые впервые выдвинули и обосновали концепцию о принципиальном различии физиологической смерти клеток (апоптоз), и их патологической гибели (некроз).
В 2002 году ученые из кембриджской лаборатории, биологи С. Бреннер, Дж. Салстон и Р. Хорвиц, получили Нобелевскую Премию по физиологии и медицине за раскрытие основных механизмов генетической регуляции развития органов и исследования программируемой клеточной смерти.
Сегодня теории апоптоза посвящены десятки тысяч научных работ, раскрывающие основные механизмы его развития на физиологическом, генетическом и биохимическом уровнях. Ведется активный поиск его регуляторов.
Особенно большой интерес представляют исследования, дающие возможность практического применения регуляции апоптоза при лечении онкологических, аутоиммунных и нейродистрофических заболеваний.
Механизм
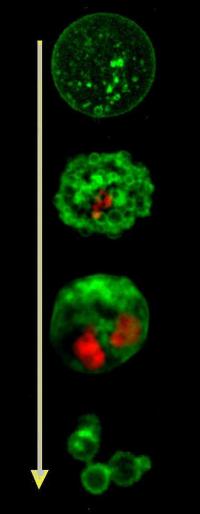 Механизм развития апоптоза на сегодняшний день до конца не изучен. Доказано, что процесс может индуцироваться малыми концентрациями большинства веществ, вызывающих некроз.
Механизм развития апоптоза на сегодняшний день до конца не изучен. Доказано, что процесс может индуцироваться малыми концентрациями большинства веществ, вызывающих некроз.
Однако в большинстве случаев генетически запрограммированная гибель клеток происходит при поступлении сигналов от молекул – клеточных регуляторов, таких как:
- гормоны;
- антигены;
- моноклональные антитела и др.
Сигналы к апоптозу воспринимаются специализированными клеточными рецепторами, которые запускают последовательные этапы внутриклеточных сложных биохимических процессов.
Характерно, что сигналом к развитию апоптоза может быть как наличие активирующих веществ, так и отсутствие некоторых соединений, препятствующих развитию запрограммированной смерти клетки.
Ответ клетки на сигнал зависит не только от его силы, но и от общего исходного состояния клетки, морфологических особенностей ее дифференцировки, стадии жизненного цикла.
Одним из базовых механизмов апоптоза на стадии его реализации является деградация ДНК, в результате чего происходит фрагментация ядра. В ответ на повреждение ДНК запускаются защитные реакции, направленные на ее восстановление.
Неудачные попытки восстановить ДНК приводят к полному энергетическому истощению клетки, что и становится непосредственной причинной ее гибели.
Механизм апоптоза — видео
Фазы и стадии
Различают три физиологические фазы апоптоза:
1. Сигнальная (активация специализированных рецепторов).
2. Эффекторная (формирование из разнородных эффекторных сигналов единого пути апоптоза, и запуск каскада сложных биохимических реакций).
3. Дегидратационная (букв. обезвоживание – гибель клетки).
Кроме того, морфологически выделяют две стадии процесса:
1. Первая стадия – преапоптоз . На этой стадии происходит уменьшение размеров клетки за счет ее сморщивания, возникают обратимые изменения в ядре (уплотнение хроматина и скопление его по периферии ядра). В случае воздействия некоторых специфических регуляторов апоптоз может быть остановлен, и клетка возобновит свою нормальную жизнедеятельность.
2. Вторая стадия – собственно апоптоз. Внутри клетки происходят грубые изменения во всех ее органеллах, однако наиболее значимые превращения развиваются в ядре и на поверхности ее внешней мембраны. Клеточная мембрана теряет ворсинки и обычную складчатость, на ее поверхности формируются пузырьки – клетка как бы кипит, и в результате распадается на так называемые апоптические тельца, поглощаемые тканевыми макрофагами и/или соседними клетками.
Морфологически определяемый процесс апоптоза занимает, как правило, от одного до трех часов.
Некроз и апоптоз клетки. Сходство и различие
Терминами некроз и апоптоз обозначают полное прекращение жизнедеятельности клетки. Однако апоптозом обозначают физиологическое отмирание, а некрозом – ее патологическую гибель.
Апоптоз является генетически запрограммированным прекращением существования, то есть по определению имеет внутреннюю причину развития, в то время как некроз происходит в результате воздействия сверхсильных внешних, по отношению к клетке, факторов:
- травма;
- ожог;
- кислородное голодание;
- недостаток питательных веществ;
- отравление токсинами и т.п.
Для апоптоза характерна постепенность и стадийность процесса, в то время как некроз наступает более остро, и четко различить стадии практически невозможно.
Кроме того, гибель клетки при процессах некроза и апоптоза отличается морфологически – первый характеризуется её набуханием, а при втором происходит сморщивание клетки, и уплотнение ее мембран.
Во время апоптоза происходит гибель клеточных органелл, однако мембрана сохраняется в целостности, так что образуются, так называемые, апоптические тельца, которые впоследствии поглощаются специализированными клетками – макрофагами или клетками-соседями.
При некрозе происходит разрыв клеточной мембраны, и содержимое клетки выходит наружу. Начинается воспалительная реакция.
Если некрозу подверглось достаточно большое количество клеток, воспаление проявляется известными с древности характерными клиническими симптомами, такими как:
- боль;
- покраснение (расширение сосудов в области поражения);
- припухлость (воспалительный отек);
- местное, а иногда и общее повышение температуры;
- более или менее выраженное нарушение функции органа, в котором произошел некроз.
Биологическое значение
3. Регуляция деятельности иммунной системы.
4. Предотвращение преждевременного старения организма.
Данный процесс играет ведущую роль в эмбриогенезе, поскольку многие органы и ткани претерпевают значительные трансформации во время эмбрионального развития. Многие врожденные дефекты возникают вследствие недостаточной активности апоптоза.
Как запрограммированная самоликвидация дефектных клеток, данный процесс является мощной природной защитой против онкологических заболеваний. Так, к примеру, вирус папилломы человека блокирует клеточные рецепторы, ответственные за апоптоз и, таким образом, приводит к развитию рака шейки матки и некоторых других органов.
Благодаря данному процессу происходит физиологическая регуляция клонов Т-лимфоцитов, ответственных за клеточный иммунитет организма. Клетки, неспособные распознавать белки собственного организма (а таких в общей сложности созревает около 97%), подвергаются апоптозу.
Недостаточность апоптоза приводит к тяжелым аутоиммунным заболеваниям, в то время как его усиление возможно при иммунодефицитных состояниях. К примеру, тяжесть течения СПИДа коррелирует с усилением данного процесса у Т-лимфоцитов.
Кроме того, этот механизм имеет большое значение для функционирования нервной системы: он ответственен за нормальное формирование нейронов, и он же может вызывать раннее разрушение нервных клеток при болезни Альцгеймера.
Одна из теорий старения организма – теория апоптоза. Уже доказано, что именно он лежит в основе преждевременного старения тканей, где гибель клеток остается невосполнимой (нервная ткань, клетки миокарда). С другой стороны, недостаточный апоптоз может способствовать накоплению в организме стареющих клеток, которые в норме физиологически отмирают, и заменяются новыми (раннее старение соединительной ткани).
Роль теории апоптоза в медицине
Роль теории апоптоза в медицине заключается в возможности поиска путей регуляции этого процесса для лечения и профилактики многих патологических состояний, вызванных ослаблением или, наоборот, усилением апопоптоза.
Исследования ведутся одновременно во многих направлениях. Прежде всего, следует отметить научные изыскания в такой значимой области медицины, как онкология. Поскольку опухолевый рост вызван дефектом генетически запрограммированной гибели мутировавших клеток, изучается возможность специфической регуляции апоптоза, с повышением его активности в опухолевых клетках.
Действие некоторых химиотерапевтических препаратов, широко применяемых в онкологии, основано на усилении процессов апоптоза. Так как опухолевые клетки более склонны к данному процессу, подбирается доза вещества, достаточная для гибели патологических клеток, но относительно безвредная для нормальных.
Также чрезвычайно важны для медицины исследования, изучающие роль апоптоза в дегенерации ткани сердечной мышцы под влиянием недостаточности кровообращения. Группа китайских ученых (Lv X, Wan J, Yang J, Cheng H, Li Y, Ao Y, Peng R) опубликовала новые экспериментальные данные, которые доказывают возможность искусственного снижения апоптоза в кардиомиоцитах при введении определенных веществ-ингибиторов.
Если теоретические исследования на лабораторных объектах удастся применить в клинической практике – это будет большой шаг вперед в борьбе с ишемической болезнью сердца. Данная патология занимает первые позиции среди причин смерти во всех высокоразвитых странах, так что переход от теории к практике трудно было бы переоценить.
Еще одно весьма перспективное направление – разработка методов регуляции данного процесса для замедления старения организма. Теоретические исследования ведутся в направлении создания программы, сочетающей повышение активности апоптоза стареющих клеток, и одновременного усиления пролиферации молодых клеточных элементов. Здесь достигнуты определенные успехи на теоретическом уровне, однако до перехода от теории к практическим решениям еще далеко.
Кроме того, масштабные научные исследования проводятся в следующих направлениях:
- аллергология;
- иммунология;
- терапия инфекционных заболеваний;
- трансплантология;
- косметология и др.
Таким образом, в недалеком будущем мы станем свидетелями внедрения в практику принципиально новых медицинских препаратов, побеждающих многие заболевания.
Использованные источники:
Апоптоз кардиомиоцитов
Активация апоптоза кардиомиоцитов.
Апоптоз — программируемая клеточная гибель, эволюционный физиологический (в отличие от некроза) механизм смерти клетки, регулирующий клеточную массу и архитектуру тканей. В норме роль апоптоза заключается в удалении поврежденных клеток и восстановлении целостности тканей. Таким образом, в здоровом организме апоптоз играет определенную адаптивную роль. В настоящее время установлена активация апоптоза кардиомиоцитов при хронической сердечной недостаточности, апоптоз утрачивает свою адаптивную роль. В результате активации апоптоза снижается количество жизнеспособных кардиомиоцитов, что снижает сократительную функцию миокарда и способствует прогрессированию сердечной недостаточности. В настоящее время хорошо известны факторы, инициирующие апоптоз кардиомиоцитов: это фактор некроза опухоли-? и другие цитокины, а также избыток оксида азота, продуцируемого кардиомиоцитами; выраженная активация перекисного окисления липидов и накопление свободных радикалов и перекисей (при этом снижается активность ферментовантиоксидантной системы — супероксиддисмутазы, кателазы, глютатионпероксидазы и токоферола); экспрессия на кардиомиоцитах рецепторов второго типа к ангиотензину-И (эти рецепторы являются активаторами апоптоза); высокая концентрация свободного кальция в цитоплазме кардиомиоцитов; катехоламины; протоонкоген с-птус. Особую роль в инициации апоптоза кардиомиоцитов играет фактор некроза опухоли-?. На поверхности кардиомиоцитов экспрессируются так называемые ?рецепторы смерти?, после связывания с которыми фактор некроза опухоли-? запускает процесс апоптоза. Связывание фактора некроза опухоли-? с рецепторами смерти и резкое усиление процессов перекисного окисления липидов, оксидативного стресса под влиянием фактора некроза опухоли вызывает активизацию в кардиомиоцитах каспазного каскада и далее под влиянием активированной каспазы-3 включается генетическая программа гибели кардиомиоцитов.
В настоящее время апоптоз кардиомиоцитов рассматривается как фундаментальный механизм, ведущий к необратимым нарушениям сократительной функции миокарда при хронической сердечной недостаточности.
Как местные, так и системные факторы могут вызывать развитие сердечной недостаточности. Сердечные факторы включают в себя повреждения миокарда (например, острый инфаркт миокарда или миокардит, развитие фиброза вследствие различных заболеваний), нарушения клапанов, аритмии (тахиаритмии или брадиаритмии), и нарушения снабжения подлежащих тканей (ишемия). Системные факторы включают любые заболевания, которые повышают содержание СО (вызывая сердечную недостаточность высокого выброса) или препятствия к выбросу (увеличение постнагрузки), такие как системная гипертензия.
Традиционное разделение на левожелудочковую и правожелудочковую недостаточность несколько условно, потому что сердце это единая система, и изменения в одной камере в итоге поражают все сердце. Тем не менее, эти термины отображают преимущественный характер поражения приводящий к сердечной недостаточности, и могут быть полезны для начальной диагностики и лечения.
Левожелудочковая недостаточность — обычно развивается в результате ишемической болезни сердца, артериальной гипертензии, аортального стеноза, большинства форм кардиомиопатий, митральной или аортальной клапанной регургитации, и наследственных заболеваний сердца (например, дефект межжелудочковой перегородки, незаращение артериального протока с большим шунтом).
Правожелудочковая недостаточность — обычно вызывается развившейся до этого левожелудочковой недостаточностью (при этом поднимается легочное венозное давление, приводящее к легочной артериальной гипертензии, и перегрузке правого желудочка) или тяжелыми легочными заболеваниями (в таких случаях это называется легочное сердце). Может вызываться также множественными легочными эмболами, заболеваниями, вызывающими закупорку легочных вен, инфарктом правого желудочка, регургитацией или стенозом трикуспидального клапана, митральным стенозом, и стенозом легочной артерии. Некоторые состояния похожи на правожелудочковую недостаточность, при том, что сердечная функция может быть в норме. Такие как перегрузка объемом и повышение системного венозного давления при полицитемии или передозировке инфузии, острая почечная недостаточность с гипергидратацией вызванной задержкой воды, или обструкцией какой-либо из полых вен.
Недостаточность обоих желудочков обычно развивается в результате поражения всего миокарда (например в результате вирусного миокардита, амилоидоза, болезни Чагаса). Недостаточность высокого выброса — развивается в результате постоянного высокого содержания СО, которая обычно развивается в результате невозможности поддерживать адекватный выброс. Состояния при которых может повышаться СО — тяжелая анемия, берибери, тиреотоксикоз, развивающееся заболевание Пажета, артериовенозная фистула, и постоянная тахикардия. СО также повышен: при различных формах цирроза, но чаще всего наблюдается снижение потоковой скорости в результате печеночных механизмов. Кардиомиопатия — это общий термин обозначающий заболевание миокарда, и иногда используется для отражения этиологии ( например ишемическая или гипертензивная кардиомиопатия). Наиболее часто, термин используется для обозначения первичного поражения миокарда желудочков, которое не вызвано наследственными анатомическими дефектами; клапанными, системными, или заболеваниями легочных сосудов; изолированными заболеваниями перикарда, или проводящей системы сердца; или заболеванием коронарных артерий эпикарда. Кардиомиопатия не всегда приводит к развитию сердечной недостаточности. Она часто идиопатическая и классифицируется как застойная дилятационная, гипертрофическая, или инфильтративно-рестриктивная кардиомиопатия.
Использованные источники:
Апоптоз сердечная недостаточность
‘> Входит в РИНЦ ® : да
‘> Цитирований в РИНЦ ® : 4
‘> Входит в ядро РИНЦ ® : да
‘> Цитирований из ядра РИНЦ ® : 1
‘> Норм. цитируемость по журналу: 2,239
‘> Импакт-фактор журнала в РИНЦ: 0,455
‘> Норм. цитируемость по направлению: 1,099
‘> Дециль в рейтинге по направлению: 2

‘> Тематическое направление: Clinical medicine
Использованные источники:
Хроническая сердечная недостаточность
Этиология.Хроническая СН, самыми частыми причинами которой в последние годы стали ИБС и инфаркт миокарда, ассоциируется, прежде всего, с нарушением систолической функции левого желудочка сердца. Среди других причин развития хронической СН следует отметить также дилатационную кардиомиопатию и ревматические пороки сердца. В старших возрастных группах (старше 60 лет) в основе развития хронической СН наряду с ИБС ведущую роль приобретает артериальная гипертензия и гипертоническое сердце, связанные, в первую очередь, с развитием диастолических нарушений. Этому способствует также возрастное уменьшение мышечного элемента и повышенное образование фиброзной ткани в миокарде пожилых. Третьей важнейшей причиной хронической СН и также в старших возрастных группах является ИНСД, который вместе с артериальной гипертензией определяет все возрастающее количество пациентов с хронической СН со сниженной систолической функцией.
Существуют факторы, способствующие прогрессированию хронической СН. Это:
— физическое и психическое перенапряжение;
— прогрессирование ИБС (развитие инфаркта миокарда, острого коронарного синдрома, нестабильной стенокардии);
— нарушения сердечного ритма (тахиаритмии – тахисистолическая форма мерцательной аритмии, трепетание предсердий, желудочковая тахикардия; брадиаритмии – синусовая брадикардия, синдром слабости синусового узла, полная атриовентрикулярная блокада);
— тромбоэмболия легочной артерии;
— резкий подъем АД, гипертензивный криз;
— воспаление легких, острые респираторные вирусные инфекции;
— почечная недостаточность (острая и хроническая);
— прием лекарственных препаратов, оказывающих кардиотоксическое действие, способствующих задержке жидкости и натрия (НПВС, эстрогены, кортикостероиды), повышающих АД, вызывающих тахикардию (изадрин, эфедрин, адреналин);
— чрезмерное употребление соли и воды;
— увеличение массы тела (особенно быстропрогрессирующее и выраженное);
— ревматическая болезнь сердца, миокардиты, эндокардиты.
Патогенез.Согласно современным представлениям, развитие хронической СН происходит по единым патогенетическим механизмам вне зависимости от этиологии повреждения и ведущую роль в этом процессе играет гиперактивация нейрогормональных систем. Она является ключевым звеном патогенеза хронической СН, ее прогрессирования, «ведет» пациента от первичного повреждения миокарда к смерти вне зависимости от характера первичного повреждения. Нейрогуморальные (нейрогормональные) изменения при хронической СН характеризуются активацией САС и снижением активности ПНС.
Активация САС на начальном этапе хронической СН оказывает определенное положительное адаптивно-компенсаторное влияние на ССС. Продолжающаяся в течение длительного времени гиперактивация САС начинает оказывать отрицательное влияние на состояние ССС и способствует прогрессированию СН вследствие следующих факторов:
— чрезмерной констрикции вен и артериол, что приводит к выраженному увеличению венозного притока (преднагрузки) и значительному росту ОПСС (постнагрузки) и снижению перфузии тканей;
— увеличения ОЦК в связи с чрезмерной активацией РААС и выраженной задержкой натрия и воды в организме (увеличенный объем циркулирующей крови значительно повышает нагрузку на миокард);
— значительного повышения потребности миокарда в кислороде вследствие избытка КА и возросшей нагрузки на миокард;
— развития тяжелых нарушений сердечного ритма (мерцательной аритмии, желудочковой тахикардии, политопной желудочковой экстрасистолии, трепетания и фибрилляции желудочков);
— непосредственного кардиотоксического эффекта (выраженная дистрофия миокарда, возможны даже некротические изменения);
— уменьшения плотности β-адренорецепторов в цитоплазматической мембране кардиомиоцитов в связи с их интернализацией путем эндоцитоза в цитозольные везикулы;
— повышения агрегации тромбоцитов и образования микроагрегатов тромбоцитов и микротромбов в микроциркуляторном русле, что ухудшает кровоснабжение тканей, в том числе и миокарда;
— перегрузки кардиомиоцитов ионами кальция вследствие активации медленных кальциевых каналов с последующей перегрузкой кальцием митохондрий; в результате резко ослабевает рефосфорилирование АДФ и наступает истощение запасов креатинфосфата и АТФ. Перегрузка кардиомиоцитов Са 2+ вызывает также активацию фосфолипаз и протеаз, которые разрушают мембрану кардиомиоцитов и вызывают их гибель;
— непосредственный кардиотоксический эффект избытка КА связывается прежде всего с перегрузкой кардиомиоцитов кальцием.
Активация РААС. Гиперактивация РААС, как и САС, на начальных этапах развития хронической СН имеет адаптивно-компенсаторное значение и направлена на поддержание гемодинамики и обеспечение перфузии органов и тканей на оптимальном уровне. Следует подчеркнуть, что адаптивно-компенсаторные реакции на начальном этапе хронической СН обеспечиваются преимущественно компонентами циркулирующей РААС. Её активация сопровождается следующими изменениями:
— повышением сократительной способности миокарда (положительный инотропный эффект);
— выраженной вазоконстрикцией (повышение тонуса вен увеличивает венозный приток крови к сердцу – возрастает преднагрузка);
— спазмированием артерий и артериол, что увеличивает постнагрузку и обеспечивает поддержание АД на должном уровне, улучшает перфузию органов и тканей;
— увеличением ОЦК за счет усиления реабсорбции натрия и воды как непосредственно под влиянием ангиотензина II, так и вследствие увеличения секреции альдостерона;
— увеличением ЧСС (положительный хронотропный эффект).
Длительная гиперактивация РААС приводит к следующим хроническим и трудно устранимым последствиям. Это:
— чрезмерное увеличение ОПСС (вследствие спазма артериол), увеличение постнагрузки, снижение перфузии органов и тканей;
— резко выраженная задержка натрия и воды (вследствие значительно увеличенной реабсорбции воды и натрия в почечных канальцах под влиянием постоянно высокого уровня ангиотензина-II и альдостерона); это приводит к значительному увеличению ОЦК, формированию отечного синдрома, увеличению преднагрузки;
— повышение чувствительности миокарда к влияниям активированной САС и КА; это сопровождается, в частности, увеличением риска возникновения фатальных желудочковых аритмий;
— повышение потребности миокарда в О2 под влиянием возрастающих постнагрузки и преднагрузки и продолжающейся активации САС;
— развитие гипертрофии, ремоделирования, апоптоза и фиброза миокарда с последующим снижением сократительной функции миокарда.
Гипертрофия миокарда и апоптоз кардиомиоцитов стимулируются ангиотензином-II, в развитии фиброза миокарда вследствие стимуляции синтеза коллагена огромную роль играет гиперпродукция альдостерона, в процессах ремоделирования миокарда участвуют одновременно ангиотензин-II и альдостерон;
— гипертрофия и ремоделирование сосудов с дальнейшим ростом ОПСС;
— хроническая клубочковая гипертензия с последующим развитием в почках фиброза, гибелью клубочков почек, падением клубочковой фильтрации, развитием ХПН различной степени выраженности;
— стимуляция секреции АДГ, который повышает реабсорбцию воды в почечных канальцах, приводит к увеличению ОЦК и способствует развитию отечного синдрома, прогрессированию СН;
— ингибирование вазодилатирующей кининовой системы (ангиотензинпревращающий фермент обладает кининазной активностью);
— нарушение функционирования системы натрийуретических пептидов. Система натрийуретических пептидов (гормонов) является основным фактором, противостоящим РААС, САС и АДГ.
При хронической СН концентрация предсердного натрийуретического пептида возрастает, однако при этом снижается выраженность натрийуретического ответа. В конечном итоге это является пусковым механизмом в прогрессировании сердечной недостаточности, приводит к развитию отечного синдрома, а в дальнейшем – к значительным гемодинамическим нарушениям.
Дисфункция эндотелия. Наиболее значительные нарушения функции эндотелия при хронической СН можно охарактеризовать следующим образом:
— увеличение экспрессии, синтеза и содержания в крови эндотелина-1, который обладает резко выраженным сосудосуживающим действием.
Эндотелин-1 провоцирует повышение ОПСС. Кроме того, эндотелин-1 участвует в развитии гипертрофии миокарда, стимулирует синтез коллагена и развитие фиброза в сердечной мышце, способствует апоптозу кардиомиоцитов;
— увеличение активности эндотелиального ангиотензин-превращающего фермента, что приводит к повышению синтеза сосудосуживающего фактора – ангиотензина-II и ускорению распада брадикинина и, следовательно, ослаблению его вазодилатирующего эффекта;
— угнетение экспрессии эндотелиальной NO-синтазы и снижение вследствие этого продукции мощного вазодилатирующего фактора – NO. Это обусловлено снижением кровотока, увеличением продукции ФНО-α (подавляет синтез NO), увеличением продукции свободных радикалов (они разрушают NO), снижением активности мускариновых рецепторов;
— возрастание прокоагулянтной активности эндотелия.
Происходит повышение уровня тромбомодулина, угнетение фибринолиза вследствие снижения продукции тканевого активатора плазминогена и увеличения продукции ингибитора активатора плазминогена;
— уменьшение продукции простациклина — важнейшего фактора, обладающего антиагрегантным и антикоагулянтным эффектами, вазодилатирующим и кардиопротективным действием (защищает миокард от ишемии);
— стимуляция выработки эндотелием супероксидных радикалов, обладающих выраженным повреждающим действием на миокард.
Кроме того, установлено, что в крови больных хронической СН повышена концентрация адреномедулина – пептида, участвующего в регуляции сосудистого тонуса и баланса жидкости и электролитов. Адреномедулин синтезируется в сосудистой стенке, в обоих предсердиях и в желудочках сердца. Повышенная концентрация адреномедулина коррелирует с клинической и гемодинамической выраженностью хронической СН. Пептид является мощным артериальным и венозным дилататором сосудов как большого, так и малого кругов кровообращения. Он вызывает почечную вазодилатацию и увеличение скорости клубочковой фильтрации, натрийурез и диурез, а также ингибирует продукцию альдостерона и эндотелина в эндотелиальных и гладкомышечных клетках.
Гиперпродукция провоспалительных цитокинов (в первую очередь, ФНО-α). Основными мишенями ФНО-α при хронической СН являются эндотелий сосудов и миоциты. Этот цитокин модулирует функцию сердца и сосудов.
Усиление процесса апоптоза кардиомиоцитов и клеток периферической мускулатуры. Апоптоз кардиомиоцитов обусловлен избыточной продукцией NO (в первую очередь под влиянием ФНО-α), увеличением концентрации ионов кальция в цитоплазме кардиомиоцитов, образованием в большом количестве свободных кислородных радикалов, увеличением содержания в кардиомиоцитах сфингизина.
Нарушение эндотелийзависимой дилатации артериол, что обусловливает длительное сохранение повышенного ОПСС и, следовательно, увеличивает потребность миокарда в кислороде и снижает его сократительную способность.
В итоге гиперактивация САС, миокардиальной РАС и РААС – АДГ при хронической СН способствует:
— раннему ремоделированию и дисфункции миокарда;
— тотальной вазоконстрикции со снижением перфузии органов и тканей;
— задержке натрия и воды, повышению ОЦК;
— повышению пред- и постнагрузки;
— дальнейшему снижению сократимости и насосной функции миокарда, прогрессированию хронической СН.
Общими чертами внутрисердечной гемодинамики при недостаточности сердца являются:
— увеличение остаточного диастолического объема крови вследствие неполной систолы;
— повышение диастолического давления в желудочках вследствие увеличения остаточного систолического объема крови;
— дилатация сердца;
— изменение МОК (уменьшение, но может быть и повышение);
— при левожелудочковой недостаточности–повышение давления в левом предсердии, малом круге кровообращения и правом желудочке (может быть отек легких), при правожелудочковой недостаточности–повышение давления в венах большого круга кровообращения, в правом предсердии.
Использованные источники:
Апоптоз — запрограммированная гибель клетки
Что такое апоптоз?
Апоптоз – физиологическая смерть клетки, представляющая собой своеобразную генетически запрограммированную самоликвидацию.
Термин «апоптоз» в переводе с греческого означает «опадающий». Авторы термина дали такое название процессу запрограммированной смерти клеток потому, что именно с ним связано осеннее опадание увядших листьев. Кроме того, само название характеризует процесс как физиологический, постепенный и абсолютно безболезненный.
У животных в качестве наиболее яркого примера апоптоза, как правило, приводят исчезновение хвоста у лягушки во время метаморфозы из головастика во взрослую особь.
По мере взросления лягушонка хвост полностью исчезает, поскольку его клетки подвергаются постепенному апоптозу – запрограммированной смерти, и поглощению деструктированных элементов другими клетками.
Явление генетически запрограммированной гибели клеток встречается у всех эукариотов (организмов, клетки которых имеют ядро). Прокариоты же (бактерии) имеют своеобразный аналог апоптоза. Можно сказать, что данный феномен характерен для всего живого, за исключением таких особых доклеточных форм жизни, как вирусы.
Апоптозу могут подвергаться как отдельные клетки (как правило, дефектные), так и целые конгломераты. Последнее особенно характерно для эмбриогенеза. К примеру, опыты исследователей доказали, что благодаря апоптозу во время эмбриогенеза исчезают перепонки между пальцами на лапках у цыплят.
Ученые утверждают, что у человека такие врожденные аномалии, как сросшиеся пальцы на руках и ногах, также возникают вследствие нарушения нормального апоптоза на ранних стадиях эмбриогенеза.
История открытия теории апоптоза
Изучение механизмов и значения генетически программируемой клеточной смерти началось еще в шестидесятых годах прошлого века. Ученых заинтересовал тот факт, что клеточный состав большинства органов на протяжении жизни организма практически одинаков, а вот жизненный цикл различных типов клеток значительно отличается. При этом происходит постоянная замена многих клеток.
Таким образом, относительное постоянство клеточного состава всех организмов поддерживается динамическим равновесием двух противоположных процессов – клеточной пролиферации (деление и рост) и физиологического отмирания отживших клеток.
Авторство термина принадлежит британским ученым – Дж. Керру, Э. Уайли и А. Керри, которые впервые выдвинули и обосновали концепцию о принципиальном различии физиологической смерти клеток (апоптоз), и их патологической гибели (некроз).
В 2002 году ученые из кембриджской лаборатории, биологи С. Бреннер, Дж. Салстон и Р. Хорвиц, получили Нобелевскую Премию по физиологии и медицине за раскрытие основных механизмов генетической регуляции развития органов и исследования программируемой клеточной смерти.
Сегодня теории апоптоза посвящены десятки тысяч научных работ, раскрывающие основные механизмы его развития на физиологическом, генетическом и биохимическом уровнях. Ведется активный поиск его регуляторов.
Особенно большой интерес представляют исследования, дающие возможность практического применения регуляции апоптоза при лечении онкологических, аутоиммунных и нейродистрофических заболеваний.
Механизм
Механизм развития апоптоза на сегодняшний день до конца не изучен. Доказано, что процесс может индуцироваться малыми концентрациями большинства веществ, вызывающих некроз.
Однако в большинстве случаев генетически запрограммированная гибель клеток происходит при поступлении сигналов от молекул – клеточных регуляторов, таких как:
- гормоны;
- антигены;
- моноклональные антитела и др.
Сигналы к апоптозу воспринимаются специализированными клеточными рецепторами, которые запускают последовательные этапы внутриклеточных сложных биохимических процессов.
Характерно, что сигналом к развитию апоптоза может быть как наличие активирующих веществ, так и отсутствие некоторых соединений, препятствующих развитию запрограммированной смерти клетки.
Ответ клетки на сигнал зависит не только от его силы, но и от общего исходного состояния клетки, морфологических особенностей ее дифференцировки, стадии жизненного цикла.
Одним из базовых механизмов апоптоза на стадии его реализации является деградация ДНК, в результате чего происходит фрагментация ядра. В ответ на повреждение ДНК запускаются защитные реакции, направленные на ее восстановление.
Неудачные попытки восстановить ДНК приводят к полному энергетическому истощению клетки, что и становится непосредственной причинной ее гибели.
Механизм апоптоза — видео
Фазы и стадии
Различают три физиологические фазы апоптоза:
1. Сигнальная (активация специализированных рецепторов).
2. Эффекторная (формирование из разнородных эффекторных сигналов единого пути апоптоза, и запуск каскада сложных биохимических реакций).
3. Дегидратационная (букв. обезвоживание – гибель клетки).
Кроме того, морфологически выделяют две стадии процесса:
1. Первая стадия – преапоптоз . На этой стадии происходит уменьшение размеров клетки за счет ее сморщивания, возникают обратимые изменения в ядре (уплотнение хроматина и скопление его по периферии ядра). В случае воздействия некоторых специфических регуляторов апоптоз может быть остановлен, и клетка возобновит свою нормальную жизнедеятельность.
2. Вторая стадия – собственно апоптоз. Внутри клетки происходят грубые изменения во всех ее органеллах, однако наиболее значимые превращения развиваются в ядре и на поверхности ее внешней мембраны. Клеточная мембрана теряет ворсинки и обычную складчатость, на ее поверхности формируются пузырьки – клетка как бы кипит, и в результате распадается на так называемые апоптические тельца, поглощаемые тканевыми макрофагами и/или соседними клетками.
Морфологически определяемый процесс апоптоза занимает, как правило, от одного до трех часов.
Некроз и апоптоз клетки. Сходство и различие
Терминами некроз и апоптоз обозначают полное прекращение жизнедеятельности клетки. Однако апоптозом обозначают физиологическое отмирание, а некрозом – ее патологическую гибель.
Апоптоз является генетически запрограммированным прекращением существования, то есть по определению имеет внутреннюю причину развития, в то время как некроз происходит в результате воздействия сверхсильных внешних, по отношению к клетке, факторов:
- травма;
- ожог;
- кислородное голодание;
- недостаток питательных веществ;
- отравление токсинами и т.п.
Для апоптоза характерна постепенность и стадийность процесса, в то время как некроз наступает более остро, и четко различить стадии практически невозможно.
Кроме того, гибель клетки при процессах некроза и апоптоза отличается морфологически – первый характеризуется её набуханием, а при втором происходит сморщивание клетки, и уплотнение ее мембран.
Во время апоптоза происходит гибель клеточных органелл, однако мембрана сохраняется в целостности, так что образуются, так называемые, апоптические тельца, которые впоследствии поглощаются специализированными клетками – макрофагами или клетками-соседями.
При некрозе происходит разрыв клеточной мембраны, и содержимое клетки выходит наружу. Начинается воспалительная реакция.
Если некрозу подверглось достаточно большое количество клеток, воспаление проявляется известными с древности характерными клиническими симптомами, такими как:
- боль;
- покраснение (расширение сосудов в области поражения);
- припухлость (воспалительный отек);
- местное, а иногда и общее повышение температуры;
- более или менее выраженное нарушение функции органа, в котором произошел некроз.
Биологическое значение
3. Регуляция деятельности иммунной системы.
4. Предотвращение преждевременного старения организма.
Данный процесс играет ведущую роль в эмбриогенезе, поскольку многие органы и ткани претерпевают значительные трансформации во время эмбрионального развития. Многие врожденные дефекты возникают вследствие недостаточной активности апоптоза.
Как запрограммированная самоликвидация дефектных клеток, данный процесс является мощной природной защитой против онкологических заболеваний. Так, к примеру, вирус папилломы человека блокирует клеточные рецепторы, ответственные за апоптоз и, таким образом, приводит к развитию рака шейки матки и некоторых других органов.
Благодаря данному процессу происходит физиологическая регуляция клонов Т-лимфоцитов, ответственных за клеточный иммунитет организма. Клетки, неспособные распознавать белки собственного организма (а таких в общей сложности созревает около 97%), подвергаются апоптозу.
Недостаточность апоптоза приводит к тяжелым аутоиммунным заболеваниям, в то время как его усиление возможно при иммунодефицитных состояниях. К примеру, тяжесть течения СПИДа коррелирует с усилением данного процесса у Т-лимфоцитов.
Кроме того, этот механизм имеет большое значение для функционирования нервной системы: он ответственен за нормальное формирование нейронов, и он же может вызывать раннее разрушение нервных клеток при болезни Альцгеймера.
Одна из теорий старения организма – теория апоптоза. Уже доказано, что именно он лежит в основе преждевременного старения тканей, где гибель клеток остается невосполнимой (нервная ткань, клетки миокарда). С другой стороны, недостаточный апоптоз может способствовать накоплению в организме стареющих клеток, которые в норме физиологически отмирают, и заменяются новыми (раннее старение соединительной ткани).
Роль теории апоптоза в медицине
Роль теории апоптоза в медицине заключается в возможности поиска путей регуляции этого процесса для лечения и профилактики многих патологических состояний, вызванных ослаблением или, наоборот, усилением апопоптоза.
Исследования ведутся одновременно во многих направлениях. Прежде всего, следует отметить научные изыскания в такой значимой области медицины, как онкология. Поскольку опухолевый рост вызван дефектом генетически запрограммированной гибели мутировавших клеток, изучается возможность специфической регуляции апоптоза, с повышением его активности в опухолевых клетках.
Действие некоторых химиотерапевтических препаратов, широко применяемых в онкологии, основано на усилении процессов апоптоза. Так как опухолевые клетки более склонны к данному процессу, подбирается доза вещества, достаточная для гибели патологических клеток, но относительно безвредная для нормальных.
Также чрезвычайно важны для медицины исследования, изучающие роль апоптоза в дегенерации ткани сердечной мышцы под влиянием недостаточности кровообращения. Группа китайских ученых (Lv X, Wan J, Yang J, Cheng H, Li Y, Ao Y, Peng R) опубликовала новые экспериментальные данные, которые доказывают возможность искусственного снижения апоптоза в кардиомиоцитах при введении определенных веществ-ингибиторов.
Если теоретические исследования на лабораторных объектах удастся применить в клинической практике – это будет большой шаг вперед в борьбе с ишемической болезнью сердца. Данная патология занимает первые позиции среди причин смерти во всех высокоразвитых странах, так что переход от теории к практике трудно было бы переоценить.
Еще одно весьма перспективное направление – разработка методов регуляции данного процесса для замедления старения организма. Теоретические исследования ведутся в направлении создания программы, сочетающей повышение активности апоптоза стареющих клеток, и одновременного усиления пролиферации молодых клеточных элементов. Здесь достигнуты определенные успехи на теоретическом уровне, однако до перехода от теории к практическим решениям еще далеко.
Кроме того, масштабные научные исследования проводятся в следующих направлениях:
- аллергология;
- иммунология;
- терапия инфекционных заболеваний;
- трансплантология;
- косметология и др.
Таким образом, в недалеком будущем мы станем свидетелями внедрения в практику принципиально новых медицинских препаратов, побеждающих многие заболевания.
Использованные источники: